INTRODUCTION
Inflammatory bowel disease (IBD), a relapsing disorder characterized by chronic idiopathic gastrointestinal (GI) tract inflammation, is categorized into ulcerative colitis (UC) and CrohnŌĆÖs disease [1]. Although environmental and genetic factors contribute to IBD progression, the exact etiology of the disease remains unclear. Currently, it is hypothesized that GI microbes may trigger a deregulated immune response in genetically predisposed individuals, resulting in chronic inflammation [2,3]. As mediators of a chronic immune response, these microbes may play a key role in the pathogenesis of autoimmune diseases, such as asthma, vasculitic diseases, and IBD [4]. The Helicobacter genus is implicated as a potential pathogenetic contributor to IBD5 and reportedly triggers IBD-like colitis in an immunocompromised murine model [5].
However, many epidemiological studies have suggested a negative association between H. pylori infection and UC [6]. Korean studies have reported an increase in the prevalence of UC with a simultaneous decrease in that of H. pylori [7]. Based on the inverse prevalence between H. pylori infection and UC, recent studies suggest a protective role of H. pylori infection against UC [8-10]. However, epidemiological studies can only suggest associations and do not establish cause-and-effect relationships. Assuming a potential protective effect of H. pylori in UC, it is reasonable to conclude that the severity of UC may be lesser in patients with than in those without H. pylori infection. However, few studies have discussed the clinical association between H. pylori infection and severity of UC.
In this study, we investigated the association, if any, between severity of UC and H. pylori infection.
METHODS
Patients
In this study, we performed a retrospective analysis of data obtained from medical records of all patients with newly diagnosed UC, who were evaluated at Korea University Ansan Hospital for H. pylori infection between January 1994 and December 2015. No patient had previously received H. pylori eradication treatment. Patients were categorized as follows: patients with H. pylori infection (HP[+] group) and those without H. pylori infection (HP[-] group). This study was approved by the Institutional Review Board of the Korea University College of Medicine (AS 16046). Informed consent was obtained from subjects.
Diagnosis of H. pylori infection
During upper endoscopy, a rapid urease test (Campylobacterlike organism test; Kimberly-Clark Ballard Medical Products, Roswell, GA, USA) or histological examination of biopsy specimens obtained from the antrum and body was performed. The biopsied tissues were fixed and stained using crystal violet for histological evaluation. A positive result on either test was diagnosed as H. pylori infection.
Assessment of disease activity of UC
The severity of UC was assessed using the following methods: 1) endoscopic severity was assessed based on endoscopic extent and endoscopic findings. Endoscopic extent was defined based on the maximum extent of the disease and was classified according to the Montreal classification [11] as follows: proctitis (inflammatory changes from the anal verge to >15 cm proximally), left colitis (extending from the rectum to the splenic flexure), and pancolitis (inflammatory changes proximal to the splenic flexure). Endoscopic findings were classified as mild (erythema, decreased vascular pattern, and mild friability), moderate (pronounced erythema, absence of vascularity, friability, and erosion), and severe (spontaneous bleeding and ulceration). 2) Symptom severity was assessed based on stool frequency, rectal bleeding severity, and a history of admission necessitated by UC aggravation. Stool frequency was classified into three groups using the Truelove-Witts score as follows [12]: mild (1ŌĆō3 stools/day), moderate (4ŌĆō6 stools/day), and severe (>7 stools/day). Rectal bleeding severity was classified based on the frequency of hematochezia as follows: minimum (no blood visible in stool), mild (blood-streaked stool <50% of the time), moderate (clearly visible blood in stool most of the time), and severe (passage of blood alone). 3) Review of details of induction therapy using sulfasalazine, 5-aminosalicylic acid (mesalazine), steroids, azathioprine, and infliximab. We concluded that patients with UC who received steroids, azathioprine, or infliximab as induction therapy had more severe UC than those who received sulfasalazine and 5-aminosalicylic acid. 4) We evaluated the severity of UC based on the Mayo score [13], which includes the following variables: stool frequency, rectal bleeding, endoscopic findings, and physicianŌĆÖs global assessment (Table 1). The estimated Mayo scores were categorized as mild disease (scores 3ŌĆō5), moderate disease (scores 6ŌĆō10), and severe disease (scores 11ŌĆō12).
Statistical analysis
All statistical analyses were performed using the IBM SPSS Statistics for Windows, version 21.0 (IBM Corp., Armonk, NY, USA). PatientsŌĆÖ mean ages were compared using the t-test, and differences in sex and endoscopic findings were compared using the chi-square test, which was also used to determine the association between H. pylori status and severity of UC. A pvalue of <0.05 was considered statistically significant.
RESULTS
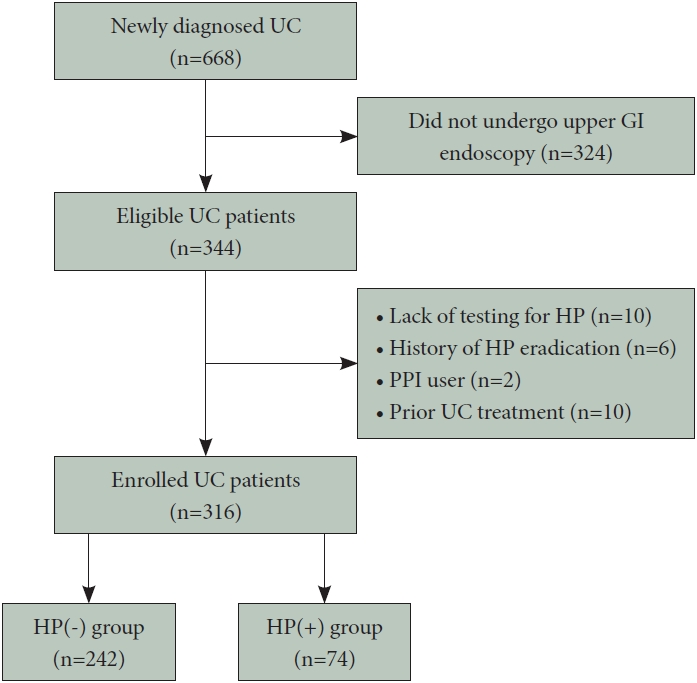
Of the 668 patients newly diagnosed with UC at our institution, 344 underwent upper GI endoscopy during the study period, and among these, 28 were excluded (10 did not undergo H. pylori testing, 6 had a history of H. pylori eradication therapy, 2 had received a proton pump inhibitor [which affects H. pylori test results] within 4 weeks, and 10 patients underwent gastroscopy after UC induction therapy). Finally, 316 patients eligible for this study underwent both upper GI and colonic endoscopic examinations. The study population was categorized into the HP(-) (n=242) and the HP(+) group (n=74) based on the results of H. pylori testing (Fig. 1). Available data showed that the H. pylori infection rate in the UC group was 23.4%.
PatientsŌĆÖ clinical characteristics
Table 2 illustrates the clinical characteristics of groups included in this study. PatientsŌĆÖ mean ages were 44.6 and 46.5 years in the HP(-) and HP(+) groups, respectively. The HP(-) and HP(+) groups comprised 140 (57.8%) and 54 (72.9%) men, respectively, and men outnumbered women in both groups; however, the difference was statistically insignificant. With regard to upper endoscopic findings, the percentage of patients with duodenal ulcers was significantly higher in the HP(+) than that in the HP(-) group (27.0% vs. 11.6%, p=0.022). We observed no statistically significant intergroup differences in other endoscopic findings (reflux esophagitis, chronic gastritis, gastric ulcers, and duodenitis).
Endoscopic severity of UC
The endoscopic extent of the disease did not show a significant intergroup difference (Table 3). The proctitis rates in the HP(-) and HP(+) groups were 28.9% and 35.2%, respectively; these differences were statistically insignificant (p=0.472). The left-sided colitis rates in the HP(-) and HP(+) groups were 46.3% and 43.2% (p=0.745), respectively, and the pancolitis rates were 24.8% and 21.6%, respectively (p=0.693).
We observed no significant intergroup difference in endoscopic findings of UC involving the colonic mucosa (Table 3). Mild disease rates in the HP(-) and HP(+) groups were 20.7% and 24.3%, respectively; these differences were statistically insignificant (p=0.635). The moderate disease rates in the HP(-) and HP(+) groups were 47.1% and 48.6% (p=0.870), and the severe disease rates were 32.2% and 27.1% (p=0.549), respectively.
Clinical symptom severity
The frequency of defecation and severity of rectal bleeding showed no significant association with H. pylori infection. Rates of admission necessitated by UC aggravation in the HP(-) and HP(+) groups were 57.9% and 59.5%, respectively, and no significant association with H. pylori infection was observed (p=0.182) (Table 4).
Induction therapy
The percentage of patients who received initial systemic steroid induction therapy in the HP(-) group was higher than that in the HP(+) group (52.5% vs. 45.9%); however, the intergroup difference was statistically insignificant (p=0.327). The percentage of patients who received azathioprine or infliximab was not statistically significantly different (Table 5).
The Mayo score
Disease severity estimated using the Mayo score did not show a significant intergroup difference (Table 6). The mild disease rates in the HP(-) and HP(+) groups were 23.1% and 21.6%, respectively; these differences were statistically insignificant (p=0.847). Moderate and severe disease rates in the HP(-) and HP(+) groups were also not statistically significantly different (56.2% vs. 45.9%, p=0.315 and 20.7% vs. 32.5%, p=0.139, respectively).
DISCUSSION
In this study, the H. pylori infection rate in patients with newly diagnosed UC was 23.4%; disease prevalence was significantly lower than that observed in the general Korean population (41.5%ŌĆō66.9%) [14,15] throughout the duration similar to our study period. This result was consistent with findings reported by previous studies, which indicates a negative association between H. pylori infection and UC [2,6,16]. Notably, the H. pylori infection status did not affect the severity of UC, including the severity of endoscopic findings, clinical symptoms, treatment regimen, and Mayo scores.
Numerous studies have investigated the effect of H. pylori infection on UC; however, these studies have reported conflicting results. Some studies suggest a protective effect of H. pylori infection against autoimmune diseases such as asthma, type 1 diabetes mellitus, and IBD [2,9,10]. Laboratory evidence, which highlights the role of H. pylori infection in immune system dysregulation, supports this impression. This finding indicates that H. pylori, through its interaction with dendritic cells, can upregulate regulatory T cells, with decreased production of pro-inflammatory cytokines, which consequently may downregulate the systemic immune response and suppress autoimmunity [2]. Another potential mechanism underlying the protective role of H. pylori infection is the differential expression of an acute and/or chronic local mucosal inflammatory response [17,18]. H. pylori may induce the production of antibacterial peptides that counteract potentially harmful bacteria implicated in the pathogenesis of IBD [17,18]. Several clinical studies have investigated the association between H. pylori infection and UC progression, which reflects disease activity [19-21]. The results of these studies show that H. pylori positivity rates were lower in patients with UC who present with pancolitis than in those with more limited disease. Reportedly, H. pylori colonization has an independent protective effect against UC disease activity.
Some other studies have reported that H. pylori infection does not protect against UC and is not associated with UC activity [1,22-24]. In these studies, the incidence of H. pylori in patients with IBD was the same as that observed in the normal population, and H. pylori infection was not significantly associated with disease extent, stool frequency, rate of admission necessitated by disease aggravation, and treatment regimen (oral or intravenous steroid therapy), which indicates a severe disease status [24,25]. However, it is important to consider the limitations of the small-scale study (42 patients) [23] and that detailed categorization for evaluation of UC activity was not reported.
In our study, we investigated the association between UC activity and H. pylori infection in a relatively large number of patients with newly diagnosed UC. Disease activity was classified in detail using various indices, including endoscopic findings, patient symptoms, treatment regimen, and Mayo scores. Endoscopically, UC is characterized by inflammatory changes observed immediately above the anorectal junction that extend proximally in a confluent and continuous manner [26]. Therefore, disease localization is easy in patients with UC. Estimation of endoscopic extent and findings is widely used to evaluate IBD severity because endoscopic severity correlates with symptom severity and is significantly correlated with histopathological findings. Stool frequency and rectal bleeding were also evaluated because these symptoms can be easily assessed and are more objective parameters compared with the patientŌĆÖs or physicianŌĆÖs global assessment. Disease activity was estimated on the basis of the induction treatment regimen. We concluded that administration of steroid, azathioprine, or infliximab as induction therapy was common in patients with severe UC and that sulfasalazine and 5-aminosalicylic acid therapy was primarily used in those with less severe disease. To avoid the possibility of spontaneous eradication of H. pylori following antibiotic therapy (particularly metronidazole), sulfasalazine, and other aminosalicylic compounds commonly used for management of UC, we only enrolled patients with newly diagnosed treatment-na├»ve UC and without H. pylori eradication treatment history.
Interestingly, in this study, we observed an inverse association between H. pylori infection and UC, which concurs with the findings of many epidemiological studies that have reported a low incidence of H. pylori infection in patients with IBD. In this study, we did not observe a protective effect of H. pylori against UC, which suggests that the inverse association may be ascribed to a coincident phenomenon and does not indicate a cause-and-effect relationship. Therefore, the lower incidence of H. pylori infection in patients with UC may be attributable to the role of other confounders such as inherent genetic or environmental factors that favor H. pylori infection in some patients and the development of IBD in some others.
Following are the limitations of this study. First, we could not address the role of potential confounders that may have affected our results. For example, low socioeconomic status may be a strong common confounder that could account for both the higher prevalence of H. pylori infection and lower prevalence of IBD observed in the study. Second, we did not investigate the effects of H. pylori eradication therapy on UC activity. Follow-up of the disease course without treatment for UC after H. pylori eradication is difficult in patients with UC, and further prospective studies are warranted to conclusively determine the exact effect of H. pylori eradication on UC disease activity. Third, we evaluated the gastric H. pylori infection status but not the H. pylori status throughout the colonic mucosa; therefore, we could not compare disease activity based on colonic H. pylori infection. Future clinical studies should investigate the effects of H. pylori on disease activity in the intestinal mucosa of patients with UC. Fourth, a selection bias cannot be excluded; H. pylori testing was not performed in all patients with newly diagnosed UC. We evaluated H. pylori infection at the physicianŌĆÖs discretion in patients who presented with dyspepsia, a history of gastric cancer, and endoscopic findings, including peptic ulcers, gastric polyps, and atrophic gastritis. Further research, including prospective cohort studies, is warranted.
In conclusion, the H. pylori infection rate in our study (23.4%) was lower in patients with UC than in the general population. Moreover, H. pylori infection did not affect endoscopic severity, symptom severity, induction therapy, or the Mayo score classification, which suggests that H. pylori infection is not associated with UC disease activity. Further prospective studies are required to conclusively establish whether H. pylori infection affects UC onset and produces chronic effects on the natural course of the disease.