흉선 유암종을 동반한 무증상의 제1형 다발성내분비선종 1예
A Case of Asymptomatic Multiple Endocrine Neoplasia Type I with Thymic Carcinoid
Article information

Trans Abstract
Multiple endocrine neoplasia type 1 (MEN1) is an autosomal dominant hereditary disorder caused by germline mutation of the MEN1 gene. It is characterized by tumors of the anterior pituitary gland, parathyroid glands, and endocrine pancreas. Thymic carcinoid tumor is uncommon and associated with a high mortality, but its natural history has not been investigated yet. We report a case of asymptomatic MEN 1 with a thymic carcinoid tumor. A 37-year-old man underwent a routine medical checkup and upper gastrointestinal endoscopy revealed a duodenal neuroendocrine tumor (NET). Further studies showed the coexistence of pancreatic tumor, parathyroid hyperplasia, pituitary adenoma, and thymoma. The patient underwent duodenal endoscopic mucosal resection, distal pancreatectomy, subtotal parathyroidectomy, and thymectomy. The pathological test revealed a duodenal NET, pancreatic NET, parathyroid hyperplasia, and thymic carcinoid tumor. He was treated for MEN 1. We report this asymptomatic case of MEN 1 with a literature review.
서 론
제1형 다발성 내분비선종(multiple endocrine neoplasia type 1, MEN 1)은 1903년 Erdheim에 의해 처음 발견되었으며, 국내 문헌에서는 1986년 첫 사례가 보고되었다[1,2]. 이는 상염색체 우성으로 유전되는 질환으로 11번 염색체의 13번 장완(11q13)에 위치하는 종양억제유전자 MEN1의 배선 돌연변이(germ line mutation)가 원인인 것으로 밝혀졌으며, 가족성 또는 산발성으로 발생하는 것으로 알려져 있다[1,3]. MEN 1은 전형적으로는 부갑상선 과증식(parathyroid hyperplasia), 장-췌장 및 십이지장의 신경내분비 종양(neuroendocrine tumor), 뇌하수체 전엽의 선종을 특징으로 하나, 부신 피질 종양 갑상선 선종, 유암종 등이 있는 경우에는 비전형적 MEN 1으로 진단할 수 있다[4].
저자들은 건강 검진 목적의 상부위장관 내시경에서 십이지장의 신경내분비 종양이 발견되어 내원한 환자에서 평가를 시행하여 췌장 신경내분비 종양 및 부갑상선 과증식, 뇌하수체 전엽선종과 흉선종을 추가적으로 발견하여 MEN 1으로 진단 및 치료한 증례를 경험하였기에 문헌 고찰과 함께 보고하는 바이다.
증 례
37세의 남자가 건강 검진에서 시행한 상부위장관 내시경검사 시 십이지장 제2부에 1 cm 크기의 상피하 종양과 함께, 갑상선 초음파에서 갑상선 주변부에 1.3 cm 크기의 종괴가 있어 추가적 평가를 위해 본원으로 내원하였다. 환자는 기저질환은 없었으며 가족력 상 아버지가 췌장 종괴로 수술 받은 병력이 있으며, 작은아버지가 췌장 종괴로 사망하였다. 신체 검진에서 특이소견은 없었으며 호소하는 증상은 없었다. 말초혈액검사에서 백혈구 9,520/mm3, 혈색소 14.3 g/dL, 혈소판 385,000/mm3였고, 혈청 생화학검사에서 aspartate aminotransferase/alanine aminotransferase 20/29 IU/L, 총콜레스테롤 232 mg/dL, 혈청요산 8.1 mg/dL (정상 2.5∼8.0 mg/dL), 칼슘 12.6 mg/dL (정상 8.5∼10.3 mg/dL), 이온화칼슘 1.6 mmol/L (정상 1.09∼1.30 mmol/L), 인산 1.7 mg/dL (정상 2.0∼4.6 mg/dL), 혈액요소질소 10.4 mg/dL (정상 6∼26 mg/dL), 크레아티닌 0.9 mg/dL (정상 0.4∼1.2 mg/dL), 가스트린 59.94 pg/mL (정상 0∼108 pg/mL)로 고칼슘혈증과 저인산혈증이 동반되었고, 부갑상선호르몬(intact parathyroid hormone, iPTH)은 121.89 pg/mL (정상 10∼57 pg/mL)로 정상의 2배 이상 증가되어 있었다. 본원에서 시행한 상부위장관 내시경검사에서 십이지장 2부에 중심부 함몰을 보이는 8 mm 크기의 점막하 종양이 관찰되며, 내시경초음파검사(endosccopic ultrasonography)에서 점막하층에 7×3 mm 크기의 균일한 저에코 종괴로 관찰되었다. 위 전정부 전벽과 후벽에 각각 4 mm, 6 mm 크기의 노란색 표면을 띄는 점막하 종양이 관찰되며, 병변의 중심부 함몰과 모세혈관의 확장소견이 관찰되었으며, 위축성 위염은 동반되지 않았다(Fig. 1). 십이지장과 위의 조직검사 결과는 모두 10개 고배율시야당 유사분열(mitosis)은 관찰되지 않았으며, Ki-67 index는 2 이하인 WHO2010 분류 Grade I의 고분화 신경내분비 종양이었다. 감별진단을 위해 시행한 컴퓨터단층촬영에서 췌장 체부에 3.5 cm 크기의 종괴, 대동맥궁 앞쪽의 가슴샘(thymus)에 1.5 cm 크기의 종괴가 관찰되며, 양측 신장에 결석이 동반되어 있었다(Fig. 2). 갑상선 초음파에서 상극(upper pole) 후방으로 좌측 1.6 cm, 우측 0.6 cm의 종괴가 관찰되며, 99mTc-sestamibi scan (99mTc-sestamethoxyisobutylisonitryl, MIBI scan) 지연영상에서 부갑상선에 방사선 섭취증가 소견이 관찰되었다. 뇌하수체 종양을 의심하여 시행한 안장 부위 자기공명영상(sella magnetic resonance imaging)에서 3 mm 크기의 결절이 관찰되었으며, 뇌하수체 기능검사는 정상으로 확인되어, 비기능성 뇌하수체 미세선종으로 확인되었다. 환자의 병력을 고려하여 MEN1 유전자 검사를 위해 동의서 작성 후 혈액을 채취하였다. 그 결과 menin 단백질을 생성하는 종양억제 유전자인 11q13에서의 여러 MEN mutation들 중 4번 엑손에서 c.784-2A>G 변이가 발견되었다.
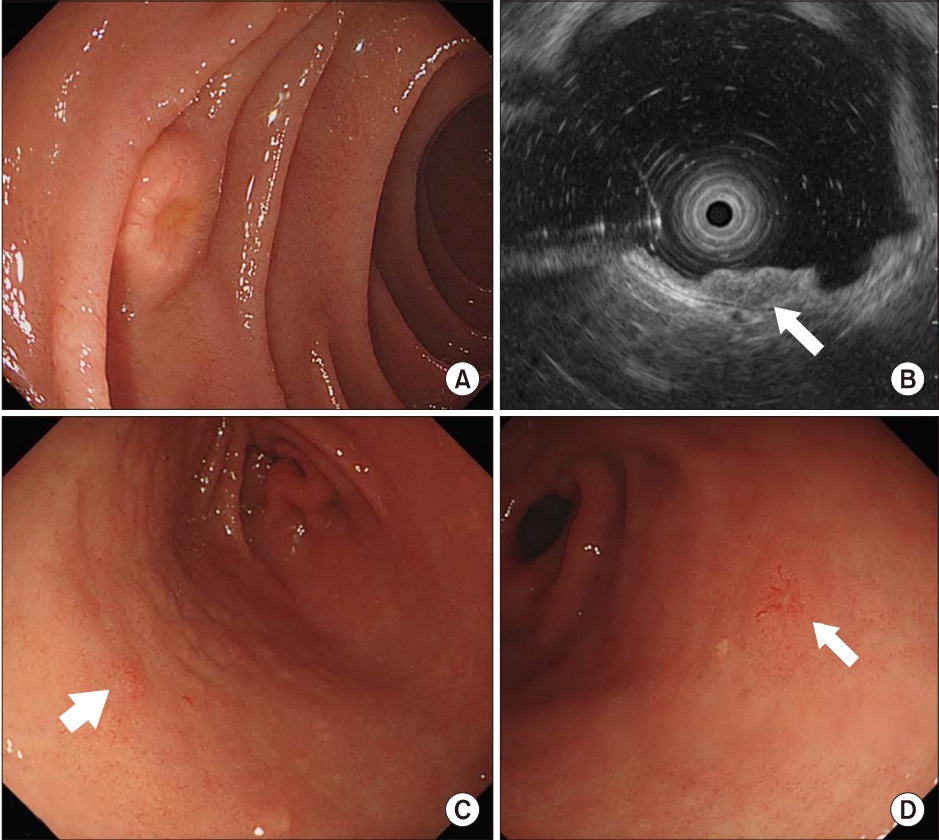
Endoscopic and endosonographic findings. (A) Endoscopic image showing a yellowish submucosal tumor with a central depression in the duodenum. (B) Endosonographic image showing a homogenously hypoechoic mass in the deep mucosal and submucosal layers (white arrow). (C, D) Endoscopic image showing additional small-sized yellowish submucosal tumors on the gastric antrum (white arrows).
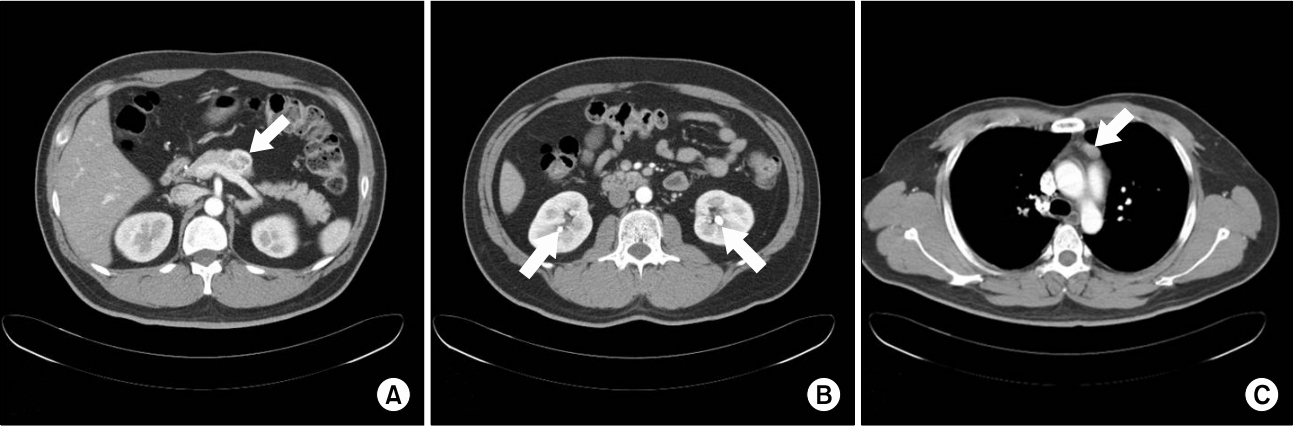
Computed tomography (CT) findings. (A) Abdominal CT image showing a cystic and solid mass with hypervascularity in the pancreatic body (white arrow). (B) Renal stones in both kidneys (white arrows). (C) Chest CT image showing a low-attenuated mass in the thymus (white arrow).
뇌하수체 선종은 비기능성 미세선종으로 추적관찰하기로 하였고, 십이지장의 내분비 종양에 대하여 내시경 점막 절제술(endoscopic mucosal resection)을 시행하였으며 절제된 병변은 0.6×0.5 cm 크기의 신경내분비 종양으로 확인되었다. 췌장종괴 및 흉선종 그리고 부갑상선 결절에 대해서 각각 외과, 흉부외과, 이비인후과와 상의하여 동시에 수술을 진행하기로 하였다. 먼저 간담도외과에서 원위췌장절제술(distal pancreactectomy)을 시행하였고, 췌장의 체부에 3.5×3.0×2.1 cm의 신경내분비 종양을 확인하였다. 이후 흉부외과에서 비디오 흉강경 흉선절제술(video-assisted thoracoscopic thymectomy)로 2.7×2.2×0.7 cm의 비전형적 유암종(atypical carcinoid)을 제거하였다. 마지막으로 이비인후과에서 좌측 상부 부갑상선의 일부를 제외한 부갑상선아전절제술(subtotal parathyroidectomy)을 시행하였고, 병리소견에서 주세포(chief cell) 의 확대와 함께 세포질이 풍부한 세포가 관찰되어 부갑상선 과증식에 합당한 소견이였다(Fig. 3).

Histological feature of each specimen. (A) Several nests of small round tumor in a well-differentiated endocrine tumor of the duodenum. Mitosis was not observed on high power field, and the Ki-proliferation index was <1% (H&E, ×200). (B) Atypical cells with mitosis visible in the well-differentiated endocrine tumor of the pancreas (H&E, ×200), (C) Thymic atypical carcinoid showing round-to-oval nuclei and fine chromatin (H&E, ×400). (D) Parathyroid hyperplasia composed of enlarged chief cells with a large amount of cytoplasm (H&E, ×400).
진단 후 약 18개월이 경과하였으며, 혈액검사와 함께 컴퓨터 단층촬영과 내시경 추적관찰 중으로, 재발 소견도 없는 상태이다.
고 찰
MEN 1은 1903년 Erdheim에 의해 처음으로 기술되었으며 반세기가 지난 1953년에 Underdahl 등에 의해 “다발성 내분비 선종”이라는 용어로 소개되었고 유전관련 질환임을 보고하였다. 이듬해인 1954년 Wermer는 이것이 단일 유전자의 돌연변이로 인한 것이며 상염색체 우성으로 유전된다고 가정하였다[5,6]. MEN 1은 부갑상선 종양, 췌장 소도세포 종양, 뇌하수체 종양 중에 2개 이상의 종양을 동반하거나, 1개 이상의 종양이 있는 환자에서 1촌 이내에 MEN 1의 가족력이 있거나, 1개 이상의 종양이 있는 환자에서 MEN1의 배선 돌연변이가 확인되었을 때 진단할 수 있다[7].
이와 비교하여 MEN 2는 제2A형, 제2B형, 제2A형의 변형인 가족성 갑상선 수질암으로 나누어 진다. MEN 2A는 갑상선수질암, 갈색세포종 및 부갑상선의 비후 또는 선종으로 구성되며, MEN 2B는 갑상선수질암, 갈색세포종과 더불어 마르판형 체형 및 점막신경종으로 구성된다. MEN 2의 경우 RET 유전자의 돌연변이로 발생하는 것으로 알려져 있다[8]. 국내에 이러한 질환의 빈도는 정확히 알려져 있지 않으나, MEN 1의 경우 대략 35,000명당 1명으로 외국에서 보고되었다[9].
MEN 1은 침범하는 장기와 기능 여부에 따라 다양한 임상양상을 보이게 된다. 부갑상선 기능항진증은 MEN 1 환자의 90% 가량에서 동반되며, 가장 먼저 발견되는 임상증상으로 알려져 있다. 대부분 선종보다는 과증식을 나타내며 생화학검사에서 고칼슘혈증이나 저인산혈증, 부갑상선 호르몬이 상승하며, 신장결석이 동반되기도 한다. 일차성 부갑상선 항진증의 경우 무증상일 경우 혈청 칼슘이 정상 상한치보다 1 mg/dL 이상 증가하거나, 사구체 여과율이 60 mL/min 이하이거나, 골밀도 검사에서 T-score가 −2.5 미만인 부위가 있거나 골절에 취약한 임상 소견이 보일 때, 나이가 50세 이하일 때 중 한가지 이상을 만족할 때 수술적 치료를 권장한다[10]. 본 증례에서는 나이가 37세이고, 혈청 칼슘이 정상 상한치보다 1 mg/dL 이상 증가하였으며, 신결석이 동반되어 수술적 적응증에 해당되었다. 또한 다발성의 과증식이 의심되는 경우에는 부갑상선 아전절제술이나 전절제술을 시행하게 된다. 부갑상선 아전절제술은 부갑상선 중 한 개 혹은 한 개의 일부인 50 mg 가량을 남기고 나머지를 모두 제거하는 방법으로 수술 후 영구적인 부갑상선 기능저하증은 1% 이하에 불과하여 전절제술과 비교하였을 때, 기능을 보존하는 데에 적합하다고 알려져 있다[11,12].
췌장 소도세포 종양은 30∼70%로 다양하게 보고된다. 그 중 가스트린 종양이 가장 흔하며 진단 당시 다발성이거나 10% 가량에서 전이성 병변이 동반된다고 알려져 있다. 인슐린종은 10∼30%에서 40세 이전의 젊은 나이에서 발견되며, 인슐린 분비로 인한 저혈당 증상이 동반될 수 있다. 그 외에 췌장 폴리펩타이드 분비 종양과 비기능성 췌장 내분비 종양 등이 있다. 췌장 소도세포 종양은 수술적 치료가 일반적이다. 췌장의 기능성 병변일 경우 인슐린, 글루카곤과 같은 호르몬 분비로 인해 합병증이 발생할 수 있으며, 비기능성 병변이라고 할지라도 악성화 가능성과 함께 주변 림프절이나 타 장기의 전이가 동반될 수 있기 때문에 1 cm 이상이거나 추적관찰 시에 크기 증가가 관찰된다면 수술을 권유하고 있다[5,13].
위의 신경내분비 종양은 그 병태생리에 따라 제1형부터 제4형까지 나눠지며, MEN 1에서는 주로 제2형이 동반되는 것으로 알려져 있다. 이러한 2형 신경내분비 종양의 특징으로는 주로 위전정부 및 저부에 1 cm 미만의 다발성 용종으로 나타나는 경우가 많고, 혈청 가스트린 수치가 상승과 함께 10∼30%에서 전이를 동반할 수 있다[14]. 위와 십이지장의 내분비 종양에 대하여 1 cm 미만의 병변에 대하여는 내시경 점막하 절제술을 시행할 수 있다고 알려져 있다[15]. 하지만 십이지장에 1 cm 미만의 내분비 종양이 있는 환자에서 위아전절제술과 근위십이지장절제술을 시행하였을 때, 십이지장에서는 14개, 위에서는 5개의 추가적인 내분비 종양과 함께, 림프절 전이가 있었다는 증례 보고가 있었다[16]. 따라서 제1형 다발성 내분비 종양 환자의 위와 십이지장 내분비 종양의 치료 방침에 대하여는 주의가 필요하다
뇌하수체 종양은 10∼50%의 MEN 1 환자에게서 보고되며, 프로락틴 분비선종이 대부분으로 알려져 있다. 원발성 뇌하수체 선종과 같은 방식으로 치료하며, 증상이 없을 경우 경과관찰하며, 증상이 있는 경우 내과적 치료를 우선으로 하고, 반응이 없을 경우 수술적 치료를 고려할 수 있다.
흉선 유암종은 MEN 1 환자의 3%에서 발견되며 기관지나 가슴샘에서 발견되며 주변 림프절 침범이 흔하다[17]. MEN 1 환자들에 대한 전향적 연구에 따르면 8년 동안 8% (85명 중 7명)의 환자들에서 흉선 유암종이 발견되었으며, 추적관찰 기간 중에 2명에서 골전이가 확인되었고, 나머지 5명에서 모두 수술 1년 후 재발이나 잔존종양이 확인되었다[18]. 따라서 흉선 유암종에 대하여 조기진단과 수술적인 치료를 하였다 하더라도 종양 자체가 빠른 성장을 보여 완치가 어려워 주의 깊은 경과관찰과 추적검사가 필요하다. MEN 1에서 부갑상선 전절제술을 할 경우, 흉선 내에 일부 남아있는 부갑상선 조직의 증식이 가능하며, 추후 흉선 유암종의 발병 가능성을 고려하여 부갑상선 절제술시 숙련된 외과의에 의한 예방적 흉선 전절제술을 함께 시행하는 것을 권장하기도 한다[17,19].
MEN 1의 임상양상은 같은 조직을 침범한 원발 종양의 임상양상과 유사하나 몇 가지 중요한 차이점을 보인다. 첫째, 여러 내분비조직에 다발성으로 발생하게 되며, 적절한 치료 후에도 재발을 흔하게 하는 편이다. 둘째, MEN 1 환자들에게서 보이는 일부 종양은 원발성으로 나타나는 종양보다 10년 이상 이른 나이에 발생하는 경향을 보이며, 특히 부갑상선 선종은 MEN 1 환자들 대부분에게서 50세 이전에 관찰된다. 셋째, 이러한 다발성 내분비 종양은 악성화 빈도가 높으며 인근의 림프절이나 원발 장기 전이가 흔하게 관찰된다. 이러한 임상적인 특징으로 인하여 치료하지 않을 경우 MEN 1 환자의 절반 가량이 50대 이전에 사망하며, 사망원인의 50∼70% 가량은 악성 종양의 진행과 관련이 있다고 알려져 있다[20,21].
MEN 1의 원인이 되는 MEN1 유전자는 11번 염색체의 장완(11q13)에 위치하며 핵단백질인 menin을 생산하며, 세포성장과 세포주기의 관리, 유전자의 안정성에 중요한 역할을 하는 종양 억제유전자로 알려져 있다. 이러한 MEN1 유전자에 불활성화 돌연변이가 생길 경우 다발성으로 종양이 발생하게 된다[22]. 유전자 검사는 DNA 배열을 이용한 연관 분석법으로 시행되는데 이전의 여러 연구를 통해 400개 이상의 변이가 발견되었다. 알려진 대부분의 돌연변이는 menin의 절단 미성숙 단백질을 생성하여 세포성장의 조절 및 세포주기의 조정에 변화를 일으켜 종양이 발생하는 것으로 보고되었다[23,24]. 특징적인 돌연변이 호발 부위는 없지만, 반복적으로 관찰되는 돌연변이로 엑손2의 c.249_252delGTCT, 엑손3의 c.628_631delACAG, 엑손10의 c.1378C>T 및 c.1546_1547insC 등 여러 돌연변이가 있으며 이 4가지 돌연변이가 전체의 약 12%를 차지하는 것으로 알려졌다[25]. 본 증례에서 발견된 4번 엑손의 c.784-2A>G 변이의 경우는 국내에서는 보고된 적이 없으며, 외국의 경우 MEN 1 관련 변이로 보고된 바가 있다(NCBI Reference Sequence: NM_001103.3). 국내 MEN 1 환자를 대상으로 한 연구에서는 엑손 7, 8에서의 돌연변이의 빈도가 높으며, 초기 진단시 부신종양이나, 폐 유암종, 위 유암종과 같은 비전형적인 증상이나 무증상이 흔하다고 보고하였다[26]. MEN 1은 상염색체 우성으로 유전되며 자녀의 50%에서 유전자 돌연변이를 가지게 된다. 따라서 MEN 1 자녀들 중 유전자 검사를 통하여 돌연변이가 없음을 확인하게 되면 불필요한 검사를 줄일 수 있다. MEN1 유전자 돌연변이가 확인된 경우에는 관련된 종양의 발생 가능성이 높기 때문에 정기적인 생화학 검사와 영상검사를 하여 적절한 치료를 하는 것이 중요하다.
요약하자면 저자들은 드문 흉선 유암종과 비전형적인 증상인 위 신경내분비 종양과 함께 부갑상선 과증식, 췌장 및 십이지장의 신경내분비 종양, 뇌하수체 전엽의 선종과 같은 3가지의 전형적인 증상이 모두 동반된 MEN 1 환자에서 각각의 병변들에 대하여 내시경적 절제와 수술적 절제를 통하여 모두 성공적으로 치료를 하였고, 새로운 MEN1 mutation을 발견하였기에 본 증례를 보고하는 바이다.
Notes
No potential conflict of interest relevant to this article was reported.